De weg naar behandeling 17
Op ons 5-jarig jubileum gaf Guy Lenaers ons een presentatie over het werk dat hij en zijn collega's aan de Universiteit van Angers (Frankrijk) verrichten. Het team van Dr. Guy Lenaers, bestaande uit 60 experts (30 medische professionals en 30 onderzoekers), is gespecialiseerd in erfelijke mitochondriale ziekten en heeft unieke expertise in het karakteriseren van mitochondriale fysiologie (de processen in het lichaam). Mitochondriën spelen een cruciale rol bij het genereren van energie (ATP) in cellen. Guy Lenaers was een van de onderzoekers die in 2000 de relatie tussen het OPA1-gen en ADOA identificeerde.
Het onderzoek omvat klinische diagnose, analyse van proeven, begrip van mitochondriale fysiologie, therapeutische benaderingen en klinische onderzoeken. Bij Autosomaal Dominante Optische Atrofie (ADOA) zijn de ogen van de patiënt gezond, terwijl de oogzenuwen zijn aangetast, wat de overdracht van visuele informatie van het netvlies naar de hersenen belemmert. De oogzenuwen werken dag en nacht; visuele informatie vormt 80% van de informatie die naar de hersenen wordt gestuurd en vereist daarom enorme hoeveelheden energie en effectieve mitochondriën. Daarom kunnen bij de meeste mitochondriale ziekten de oogzenuwen disfunctioneren. De effecten variëren van patiënt tot patiënt, zelfs als dezelfde mutatie individuen in dezelfde familie beïnvloedt.
Ondanks het feit dat het OPA1-gen meer dan 20 jaar geleden is geïdentificeerd, is het ontwikkelen van gentherapie zeer uitdagend. De belangrijkste redenen hiervoor zijn:
1. Er zijn meer dan 400 mutaties in het OPA1-gen
2. Er zijn twee verschillende ziekteveroorzakende mechanismen geassocieerd met OPA1-varianten, haplo-insufficiëntie en dominant negatieve effecten
3. OPA1 genen coderen voor 8 verschillende eiwitten met verschillende functies in de mitochondriën
4. Te veel OPA1-expressie is ook giftig.
Ten eerste maken de 400 mutaties het moeilijk om gewoon het gen te bewerken, omdat dit zou vereisen dat bijna alle families afzonderlijke behandelingen ontvangen. Ten tweede, hoewel het mogelijk is om met een haplo-insufficiëntie om te gaan door OPA1 aan te vullen, is de ziekte soms dominant negatief, wat betekent dat het gemuteerde gen een negatief effect heeft op het normale gen. Ten derde richten sommige behandelingsbenaderingen zich op het herstellen van één OPA1-functie, terwijl de andere functies niet worden aangepakt. Ten vierde is het eenvoudig injecteren van veel OPA1 giftig en kan het minstens zoveel schade aanrichten als het ontbreken van OPA1.
De mogelijke oplossing voor dit probleem in Angers is het gebruik van een aanpak genaamd trans-splicing. Wanneer het OPA1-gen beschadigd is, maakt het ook beschadigd pre-RNA (een tussenstap tussen DNA en RNA), wat leidt tot pathogene OPA1-eiwitten. Trans-splicing snijdt het beschadigde deel van het pre-RNA af en vervangt het door een deel dat normaal functioneert. Deze aanpak kan worden gebruikt voor het overgrote deel van de 400 mutaties; op dit moment kan ongeveer 80% van de mutaties worden gecorrigeerd, wat overeenkomt met 90% van de patiënten die mogelijk met deze aanpak kunnen worden behandeld. Bovendien lost het ook de andere drie problemen met betrekking tot OPA1-behandelingen op. Momenteel bevindt deze aanpak zich in het basisonderzoeksstadium, met nog wat werk dat in het laboratorium moet worden gedaan, en de veiligheid van de aanpak moet worden getest op apen. Voor de ontwikkeling van deze daadwerkelijke behandeling is een partnerschap met een farmaceutisch bedrijf nodig dat een aanzienlijk bedrag (vele miljoenen euro's) investeert voor het uitvoeren van klinische studies. Als dergelijke behandelingen worden ontwikkeld, betekent dit niet dat alle patiënten er daadwerkelijk baat bij zullen hebben, aangezien deze vormen van therapieën een aanzienlijke prijs hebben en gezondheidssystemen mogelijk niet bereid zijn om voor deze therapieën te betalen, zelfs na ontwikkeling.
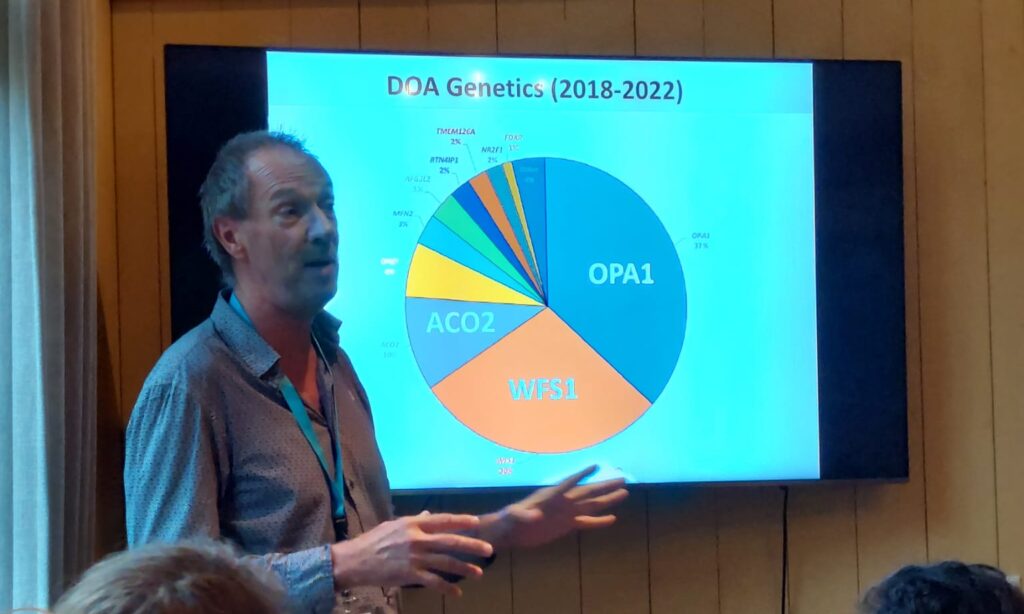
Sinds 2017 ontdekte Guy Lenaers en zijn team dat slechts ongeveer een derde van de nieuwe ADOA-patiënten een OPA1-mutatie heeft, terwijl een groot aantal andere genen is geïdentificeerd als gerelateerd aan optische atrofie, waarbij de genen WFS1 en ACO2 de tweede en derde belangrijkste zijn. Momenteel richten bijna alle geplande RNA- en DNA-therapieën zich uitsluitend op het OPA1-gen. Dit betekent dat onderzoek naar de behandeling van andere mutaties moet worden gestart.
Een andere benadering betreft neuroprotectoren, zoals vitamine B3 (ook wel nicotinamide genoemd), spermidine en taurine. Neuroprotectoren zijn een meer algemene methode om de visuele functie te behouden en zijn niet afhankelijk van mutaties. Momenteel is het team van Guy Lenaers betrokken bij een onderzoek naar de veiligheid van B3, waarvan de resultaten in 2025 beschikbaar zouden moeten zijn. Om de effectiviteit van deze aanpak verder aan te tonen, zou een dubbelblinde klinische studie met minstens 150 patiënten moeten worden opgezet. Echter, B3 is een vrij verkrijgbaar geneesmiddel, er is momenteel geen farmaceutisch bedrijf dat het financiert voor klinische proeven. Een dergelijke proef zou meer dan 1,0 miljoen euro kosten en is volledig afhankelijk van publieke financiering. Tegelijkertijd, als de therapie werkt, vanwege de lage kosten van vitamine B3, zijn gezondheidssystemen mogelijk meer bereid om voor deze behandeling te betalen.